Cristiane
Boé e Ulysses Fagundes Neto
Introdução
A
função pancreática normal garante efetiva digestão e absorção dos nutrientes. Vale
ressaltar que o pâncreas adquire plena maturidade funcional na produção
enzimática a partir dos 2 anos de idade (Figuras 1-2-3).

Figura
1- Localização anatômica do pâncreas no interior da cavidade abdominal.

Figura
2- Anatomia e histologia do pâncreas e sua localização na segunda porção do
duodeno.
Por
outro lado, as manifestações clínicas da insuficiência pancreática (IP) ocorrem
quando as secreções exócrinas do pâncreas não mantêm uma função digestiva
normal, afetando a qualidade de vida e, como consequência, pode resultar em desnutrição.
A
principal causa de IP é a pancreatite crônica, que apresenta a estimativa de
afetar 0,4% a 5% da população mundial. Entretanto, em crianças, a causa mais
comum é a fibrose cística (FC). A prevalência de IP exócrina nos casos de
pancreatite crônica é de 30 a 40%, enquanto que na FC é de 80 a 90% (Figura C).
Desde
1938, após a aprovação pelo Food and Drug Administration (FDA), a reposição de
enzimas pancreáticos tem sido a principal ação terapêutica para aqueles
pacientes que sofrem de má absorção dos nutrientes secundária à insuficiência
pancreática. O nível das enzimas pancreáticas secretadas no intestino delgado varia
segundo a quantidade e o tipo de lipídios ingeridos durante uma refeição, além
de também apresentar uma oscilação individual. Em virtude da existência destas
variáveis, determinar o nível exato de enzimas secretadas por um indivíduo
normal é muito difícil.
Etiologias
As causas
de IP em crianças podem ser divididas em 2 categorias:
- hereditárias
- adquiridas
A
maioria das causas hereditárias faz parte de doenças sistêmicas, como
explicitado no quadro 1, abaixo.

Quadro
1- Causas de IP na infância.
Apresentação
Clínica da Insuficiência Pancreática
O
diagnóstico é em grande essência clínico. No caso de um determinado paciente ser
portador de alguma causa conhecida de IP que se apresenta com perda de peso e
esteatorreia, está indicado o início do tratamento sem que seja necessária a
realização de extensa investigação laboratorial. A IP se instala quando restar
menos de 10% da função pancreática exócrina.
Esteatorreia
(fezes espumantes, malcheirosas que boiam no vaso sanitário), perda de peso,
desconforto e distensão abdominal são sintomas comuns relacionados com a
digestão inadequada dos lipídios.
Inicialmente,
a digestão de proteínas e amido se mantém adequada. Com a progressão da IP a má
absorção de lipídios leva ao quadro de desnutrição, má absorção de vitaminas
lipossolúveis (A, D, E, K), depleção de micronutrientes e diminuição de
lipoproteínas circulantes.
A IP
exócrina pode por si só, de forma isolada, causar ou exacerbar transtornos da
motilidade do trato digestivo. Nestas circunstâncias, há alteração na regulação
neuro-hormonal, especificamente na produção de colecisticinina e polipeptídeos
pancreáticos (afetados pelo alimento não digerido no intestino), levando ao
esvaziamento rápido do estômago e alterando a motilidade antroduodenal e da
vesícula biliar.
Nos
pacientes, nos quais o tratamento ainda não se iniciou, é notório o fato de que
se alimentam em curtos espaços e que apresentam tempo de trânsito no intestino
delgado mais rápido, cujos sintomas podem ser revertidos com o emprego de
enzimas pancreáticos exógenos.
Diagnóstico
A caracterização
diagnóstica pode ser estabelecida por métodos diretos e indiretos, a saber:
Indiretos: são
menos invasivos, mas que apresentam alta sensibilidade e especificidade, tais
como:
- determinação quantitativa da eliminação
de gordura nas fezes, as quais devem ser coletadas por um período de 72 horas,
pelo método de Van de Kamer;
- elastase fecal, dosada pela técnica ELISA,
e que avalia exclusivamente a produção enzimática endógena;
- quimiotripsina fecal, determinada por método
colorimétrico, avalia a produção de ambas enzimas, endógenas e exógenas,
portanto, pode apresentar resultados duvidosos em pacientes em vigência de terapia
de reposição enzimática.
Diretos (quantitativos)
- teste da secretina-ceruleina;
- teste da secretina-pancreozimina (padrão-ouro, porém invasivo, sem valores de
referência bem definidos em crianças).
¡ Teste
da secretina-pancreozimina
Deve-se
utilizar sonda gastroduodenal de dupla via posicionada sob controle
fluoroscópico; uma das vias drena o conteúdo gástrico e a outra o duodenal.
Secreções duodenais são coletadas em intervalos de 10 minutos, em três períodos
após injeção intravenosa de secretina e em três períodos após infusão
intravenosa de pancreozimina. Os aspirados são avaliados quanto ao volume,
medida do pH, concentração de bicarbonato, amilase, tripsina, lipase,
bilirrubinas, fosfatase alcalina, cálcio e magnésio. Amostras são encaminhadas
para microscopia e citologia. A ocorrência de produção enzimática reduzida após
injeção de pancreozimina corresponde ao índice mais sensível para avaliar a
diminuição da função pancreática.
Principais
Enfermidades Causadoras de Insuficiência Pancreática
Fibrose Cística
Trata-se
de doença de herança autossômica recessiva que afeta múltiplos tecidos
secretores epiteliais cuja incidência é de 1:3000 nascidos vivos em indivíduos
caucasianos (Figura 3).

Figura
3- Tipo de herança da FC.

Figura
4- Principais manifestações clínicas e órgãos afetados pela FC.
As
manifestações clínicas da FC são reflexo de mutações no gene CFTR (cystic
fibrosis transmembrane condutance regulator). Geralmente ocorrem 2 mutações
(forma clássica) ou 1 mutação (forma não-clássica), sendo a mais frequente aquela
que afeta o gene ΔF508.
Estas mutações no gene CFTR resultam em transporte anormal de sódio e cloro. Em
condições normais, o cloro intraluminal é trocado por bicarbonato, o que torna
o ambiente alcalino, permitindo, desta forma, que altas concentrações de
proteínas fiquem em seu estado solúvel. Em virtude da mutação no gene, o
resultado é a presença de uma secreção viscosa e ácida, levando à obstrução
ductal. Esta obstrução resulta em destruição tecidual por retenção de enzimas
proteolíticas, fibrose, substituição gordurosa, formação de cistos e
insuficiência pancreática exócrina.

Figura
5- Esquema representativo comparando a glândula sudorípara de um indivíduo
normal com a do portador de FC.
O
mesmo ocorre com as secreções pulmonares (doença pulmonar crônica com infecções
bacterianas recorrentes) e infertilidade no sexo masculino devido a azoospermia
obstrutiva.
Nos
recém-nascidos deve-se suspeitar do diagnóstico de FC na presença de íleo
meconial, prolapso retal, “suor salgado”, icterícia prolongada, anemia
hemolítica sem causa aparente, hipoalbuminemia e edema.
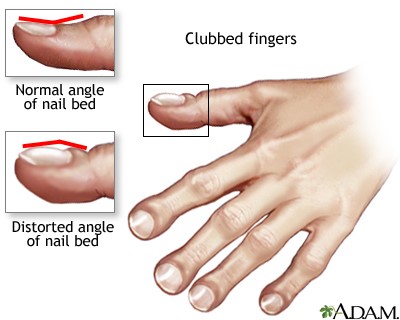
Nas
crianças maiores a existência de dedos em baquetas de tambor, pólipos nasais,
assim como secreção do trato respiratório colonizada por S. aureus ou P.
aeruginosa, são achados importantes.
Triagem
Neonatal
Desde
2001 o Ministério da Saúde criou o Programa Nacional de Triagem Neonatal,
incluindo o diagnóstico precoce para FC, anemia falciforme e outras
hemoglobinopatias.
Realiza-se
rotineiramente a dosagem sérica do tripsinogênio imunorreativo (IRT), indicador
indireto da doença pois avalia a integridade da função pancreática (Tabela 1).
Caso
a dosagem revele-se positiva (>70ng/ml), deve-se realizar nova dosagem após
15 dias, e se o teste permanecer positivo, deve-se realizar teste genético ou
teste do suor.
O Íleo
meconial pode estar relacionado a testes falso‐negativos, pois com a desobstrução intestinal ocorre rápida queda dos níveis de IRT no sangue. Mesmo diante de
um IRT normal, o que não descarta completamente o diagnóstico de FC, se uma
criança apresentar sintomas sugestivos da doença, deve ser submetida ao teste
do suor.


Nenhum comentário:
Postar um comentário